Liquid biopsy and the analysis of circulating tumor DNA (ctDNA) play an increasingly important role in cancer care and drug development by providing diagnostic and prognostic information, guiding treatment selection, and assessing response to therapy. Additionally, research has demonstrated the promise of liquid biopsy as a complementary approach to tissue-based methods that can be potentially transformative, improving turnaround times, reducing logistical complexity, and lowering costs. More recently, DNA methylation analysis in the blood, powered by advances in wet-lab chemistry and bioinformatics, has enabled the use of epigenomic profiling to enhance the sensitivity of liquid biopsy, providing a deeper understanding of tumor biology and expanding the promise of precision oncology beyond what genomic information can offer.
What DNA Methylation Tells Us
DNA methylation is a chemical modification of DNA involving the transfer of a methyl group to the C5 position of cytosine to form 5-methylcytosine, a phenomenon that often occurs in the promoter and enhancer regions of a gene and is a key contributor to gene regulation.
Aberrant DNA methylation is a hallmark of cancer. Global hypomethylation leads to oncogene activation or chromosomal instability. Hypermethylation in promoter regions can silence tumor suppressor genes. Additionally, methylation patterns of ctDNA are consistent with the tumor cells or tissues from which they originate.
By uncovering tumor-specific promoter hypermethylation and global methylation patterns, testing for DNA methylation in the blood adds actionable epigenomic information complementary to genomic analysis — information that can be used to support both drug development and patient care. Key plasma-based epigenomic applications include biomarker discovery, assessment of early molecular response, longitudinal monitoring, and minimal residual disease (MRD) detection, all at a level of sensitivity that offers new insights that support clinical oncology research. Importantly, DNA methylation has been shown to be less impacted by clonal hematopoiesis of indeterminate potential, age, and the presence of comorbidities including asthma, COPD, and rheumatoid arthritis.
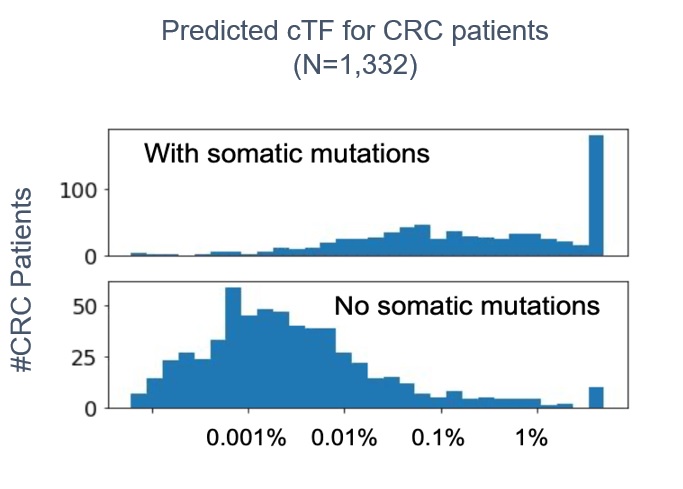
Methylation signals can be detected and measured in the blood from patients with little or no tumor shedding. In a study of 559 lung, breast, and colorectal cancer patients and 131 healthy donors, a new liquid biopsy panel that detects a sample-wide DNA methylation signal demonstrated strong analytical performance (see figure 1). The GuardantInfinity panel detected tumors in cancer patient plasma samples across the tumor types studied with 91 percent sensitivity while detecting no tumors in plasma samples from healthy donors (97 percent specificity).
Overcoming Technological Challenges of DNA Methylation Analysis
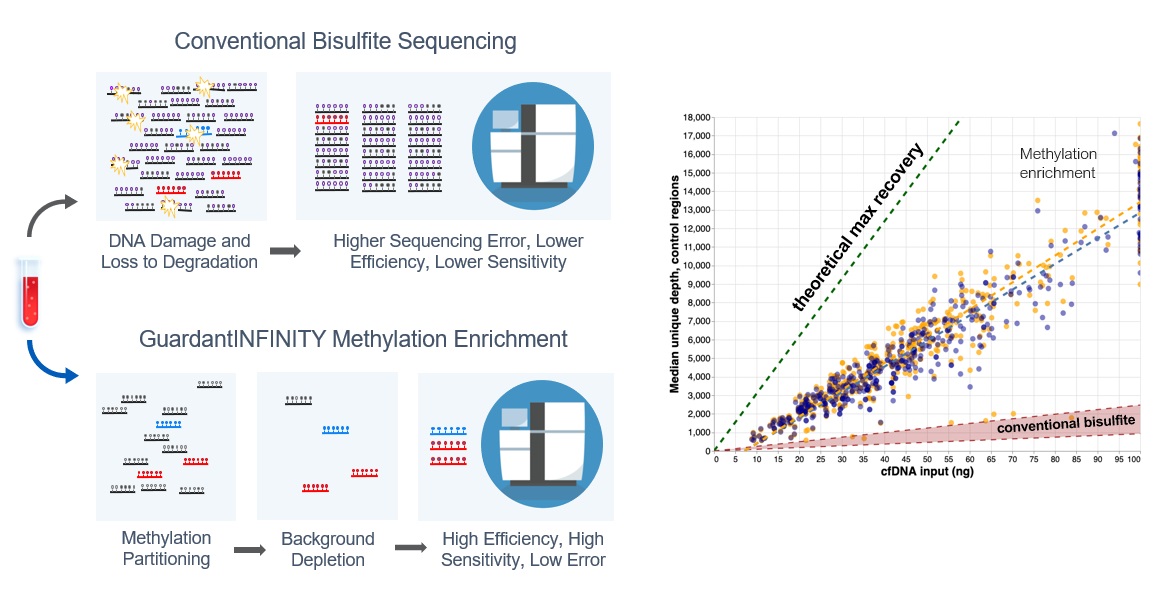
The key to a highly efficient, sensitive, and scalable sequencing method for broadly capturing methylation data is the non-destructive enrichment of methylated DNA molecules. This requires avoiding the application of harsh chemicals, such as conventional bisulfite treatments prior to sequencing, which can cause significant DNA damage and loss to degradation, resulting in higher error rates and lower sensitivity. Improving the methylation enrichment process allows lower DNA inputs and significantly increases the molecule recovery, thus improving assay sensitivity and specificity, which make early detection and more reliable quantitation possible, even in patients with low tumor shedding (see figure 2). These advantages are particularly beneficial when profiling the epigenome from blood samples that often have limited DNA availability due to low shedding tumors.
Just as important as a highly efficient methylation sequencing process is the addition of a robust bioinformatics strategy to analyze the sequencing data. One critical factor to consider is the number of methylated regions interrogated by the assay. Current genomic assays typically survey hundreds of cancer-related genes for somatic alterations of various classes; adding 15 MB of epigenomic coverage can significantly improve the sensitivity and precision of ctDNA detection. A second factor to consider is constraining the profiling to sites specific to cancer — i.e., training the panel to identify specific tumor types by profiling tumor-specific plasma samples — which can improve sensitivity, specificity, and process efficiency. Third, the ability to identify promoter hypermethylation in dozens of genes related to homologous recombination repair, processes related to immunotherapy, and important tumor suppressor genes may yield actionable information.
DNA Methylation Use Cases
BRCA1 promoter methylation in clinical management. A study in Nature Communications by Dominik Glodzik and colleagues showed that hypermethylation of the BRCA1 gene promoter was highly prevalent in triple-negative breast cancer (TNBC), associated with loss of BRCA1 mRNA expression, and showed higher levels of BRCA1 promoter methylation in matched peripheral blood DNA than patients with unmethylated tumors and no BRCA1/2 variants. Patients with BRCA1 promoter hypermethylation had similar molecular and clinical features as those with BRCA1-inactivated tumors, including comparable clinical outcomes following adjuvant chemotherapy. Glodzik notes that promoter methylation occurs in 16–57 percent of TNBC patients and is twice as frequent as BRCA1 pathogenic variants in early-stage TNBC (24 percent vs. 10.5 percent, n=237). In a study by Maha Elazezy and team published in Molecular Oncology in patients with ovarian cancer, the status of BRCA1 promoter hypermethylation in plasma was independently correlated to longer relapse-free survival. These studies demonstrate the clinical utility of BRCA1 promoter methylation and its influence on response to therapy in both breast cancer and ovarian cancer.
Minimal residual disease detection. The presence of residual ctDNA after curative intent treatment can identify patients with MRD and predict recurrence. Incorporating epigenomic signatures and tracking thousands of cancer-specific methylation features using a methylation-informed liquid biopsy assay enables highly sensitive and specific MRD detection in certain cancers. A study of 103 colorectal cancer patients who received curative-intent surgery demonstrated favorable sensitivity and specificity of the Guardant Reveal ctDNA assay for MRD detection — performance comparable with tumor-informed approaches. This makes it possible to identify patients who may benefit from additional systemic therapy and to avoid unnecessary therapy for lower-risk patients, even when tumor tissue is not available or sufficient for MRD detection.
Methylation status as a predictive biomarker for immune checkpoint inhibition. A comprehensive survey of 179 DNA repair genes demonstrated that promoter hypermethylation of DNA repair genes correlated with a previously identified interferon-related gene signature that predicted response to anti-PD-1 therapy in melanoma and head and neck squamous carcinoma (HNSCC) cancer patients. The same study identified a significant positive correlation between the methylation status of DNA repair genes and the expression of these same inflammatory genes related to this predictive signature in HNSCC. These findings suggest that immunotherapy treatments may be combined with DNA hypomethylation agents to further explore these correlations to improve the efficacy of immune checkpoint inhibition agents.
Harnessing the Power of Epigenomics
DNA methylation was first described decades ago, yet DNA methylation analysis has only recently emerged as a powerful and pragmatic tool for deepening our understanding of previously untapped epigenomic elements of the individual cancer patient’s tumor and using these insights to guide drug development and clinical management. By combining highly efficient, non-destructive methylation sequencing technology with advanced bioinformatic tools, DNA methylation analysis is poised to play an increasingly significant role in clinical research. Liquid biopsies already provide faster time to result and reduced dependence on tissue samples. The addition of epigenomic analysis to a genomic liquid biopsy assay greatly increases its ability to identify novel biomarkers and complex methylation signatures to accelerate precision oncology from a single plasma sample